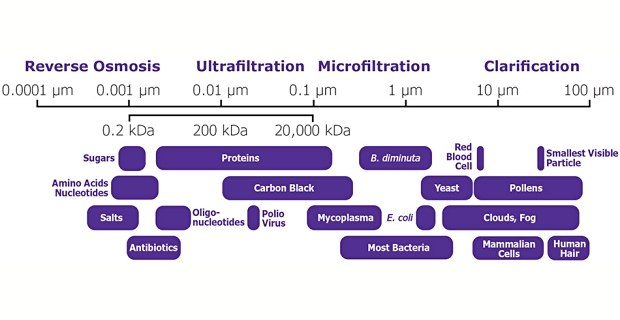
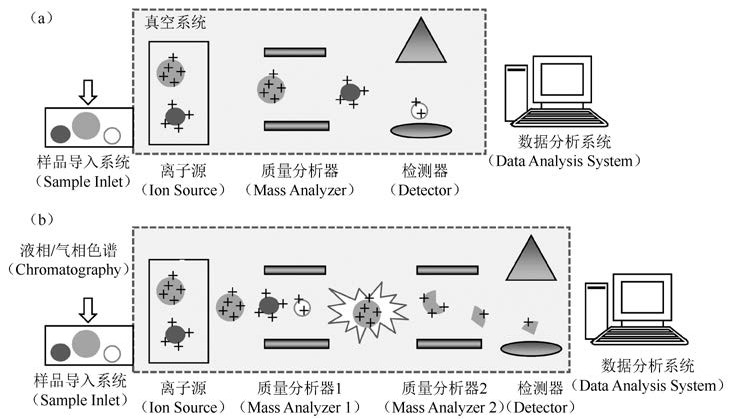
我开始更新公众号了
我在 2017 年或者更早,就搭建了自己的独立博客,那时主要写一些随笔,更的很少很慢。研究生和工作之后,开始转向写一些技术内容,当然了,主要是一些学习笔记,毕竟我还没有到能独立输出知识的地步。同时博客的更新频率也相对多了些。一方面,我关注的方向比较多,涉猎广泛杂乱;另一方面,我逐渐养成整理知识体系的习惯,并使用了一些 Markdown 类的管理工具,方便转成博客。
属于独立博客的时代已经过去了。我所坚持的似乎是一件很老套的事情,就如同在精心维护一片野花园,却处在偏远山中,少有人游览。但我觉得呢,那些花儿本身,就值得被栽种盛开。
我想,写博客的目的,首先是记录。“那些很渺小的东西,都是我人生的大事。”我始终保持对文字的敬畏,我们应该记录和表达一些东西。只可惜,我好像能静下来思考的时间越来越少,我好久没能写一篇完整的随笔。
其次,写博客的目的是共享。我很乐意分享我学习和总结到的一些东西,并期待在提供帮助的同时,也能收到一些反馈。从后台统计看,我很高兴一些内容似乎帮助到了某些陌生人,至少,对 AI 起了帮助。
此外,我认为比无知更可怕的是误解。我担心在传递这些内容的同时,由于我的不足也传递了谬误。所以,亲爱的陌生人,如果您有任何的疑问,请一定不要吝惜与我交流( jiangshen@outlook.com )。