使用血球计数板进行细胞计数
血球计数板
血球计数板用优质厚玻璃制成,通过 H 形凹槽分为两个相同的计数池。每个计数池的两侧都有一个支持柱,上面覆盖着特制的盖玻片,形成高度为 0.10 mm 的计数池。
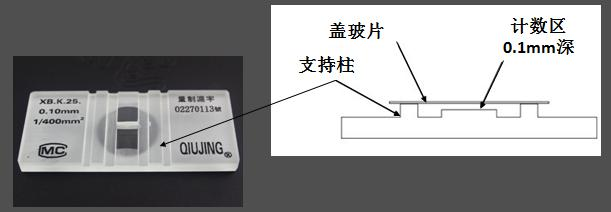
计数池内划分为长和宽各为 3 mm 的方格,总共分为 9 个大方格。每个大格面积为 1.0 mm,体积为 0.1 mm3,即一个大方格的体积是 0.1 μL。
血球计数板用优质厚玻璃制成,通过 H 形凹槽分为两个相同的计数池。每个计数池的两侧都有一个支持柱,上面覆盖着特制的盖玻片,形成高度为 0.10 mm 的计数池。
计数池内划分为长和宽各为 3 mm 的方格,总共分为 9 个大方格。每个大格面积为 1.0 mm,体积为 0.1 mm3,即一个大方格的体积是 0.1 μL。
| 试剂 | 用量 | 终浓度 |
|---|---|---|
| 10X Tris-甘氨酸缓冲液 | 100 mL | 1X |
| 甲醇 | 200 mL | 20% (V/V) |
| 去离子水 | to 1000 mL |
甲醇能减少凝胶的膨胀并提高蛋白质与硝酸纤维素膜的结合效率。转膜效率受以下因素影响:电泳缓冲液中是否存在 SDS、转膜缓冲液的 pH 及转膜前凝胶中的蛋白质是否已染色。为最大限度地提高转膜效率,SDS 的浓度不应超过 0.1%,转膜缓冲液 pH 必须 ≥8.0。
溴氯丙烷是一种可取的氯仿替代物,因为它毒性更小且不易挥发。此外,溴氯丙烷可以提供更好的液相分离,从而更好地从总 RNA 中去除基因组 DNA,获得更纯的 RNA。
Table 1. Relevant physiological parameters for different animals1
| Animal species | Weight (kg) | Blood volume (mL) | Blood flow (mL/min) | Total surface area (m^2) | Life-span (year) | Normal oral (mL/kg) | Normal i.v. (mL/kg) |
|---|---|---|---|---|---|---|---|
| Mouse | 0.02 | 1.7 | Liver: 1.8; kidney: 1.3 | 0.008 | 2.7 | 10 | 5 |
| Rat | 0.25 | 13.5 | Liver: 13.8; kidney: 9.2 | 0.023 | 4.7 | 5–10 | 2.5 |
| Rabbit | 2.5 | 165 | Liver: 177; kidney: 80 | 0.17 | 8.0 | 5–10 | 1–2 |
| Rhesus monkey | 5 | 367 | Liver: 218; kidney: 138 | 0.32 | 22 | 5–8 | 0.5–1 |
| Dog | 10 | 900 | Liver: 309; kidney: 216 | 0.51 | 20 | 5–8 | 0.5–1 |
Table 2. Recommended Dose Volumes for Common Laboratory Animals2
反应结束后需要确认扩增曲线及熔解曲线,标准扩增曲线为 S 型,熔解曲线呈单峰;
Ct 值介于 20–30 之间时,定量分析最准确;Ct 值小于 10 时,需要将稀释模板后,重新进行实验;Ct 值介于 30–35 之间时,需要提高模板浓度,或者增大反应体系的体积,以提高扩增效率,保证结果分析的准确性;Ct 值大于 35 时,检测结果无法定量分析基因的表达量,但可用于定性分析。
定量实验至少需要三个生物学重复;所有复孔间的差别应该在 0.5 Ct 内;在 35 个循环以后,Ct 值变化较大,定量结果可能不太可靠。
标准扩增曲线为 S 型,如下图:
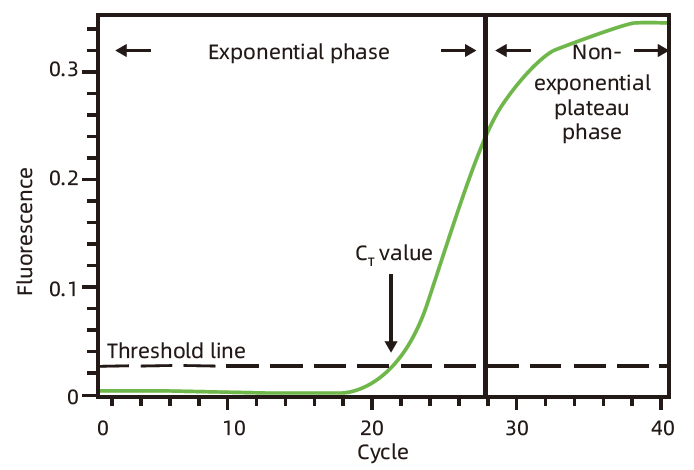
PCR 前期,目的 DNA 以指数形式扩增,这一扩增时期称为指数期(Exponential phase);PCR 后期,由于引物和 dNTP 等反应底物的减少,目的 DNA 片段扩增的速度变慢,直至停止,这一扩增时期称为平台期(Plateau phase)。qPCR 过程中,荧光值随着目的 DNA 的扩增而增加,当荧光值达到阈值时的循环数称为 Ct 值,Ct 值越小,说明目的 DNA 的初始浓度越大。
It contains a 5X pre-mixed reagent containing all of the components needed for quantitative RT-PCR reverse transcription (PrimeScript RTase, RNase Inhibitor, Random 6 mers, Oligo dT Primer, dNTP Mixture, and reaction buffer), and a reaction can be started simply by adding template RNA and water.