AutoDock 教程
1 关于 AutoDock
AutoDock 是一个预测小分子(如底物或候选药物)如何与已知 3D 结构的受体结合的分子对接工具。目前主要有 AutoDock 4 和 AutoDock Vina 两种发行版本,其中 AutoDock 4 还有一个 GPU 加速版本——AutoDock-GPU。1
AutoDock 4 进行分子对接时由两部分组成:
autogrid 计算受体的 Grid;
autodock 执行配体与靶蛋白 Grid 的对接。
注:AutoDock Vina 不需要选择原子类型及预先计算 Grid,Grid 在执行对接时计算,因此能够用于虚拟筛选(批量对接);且 AutoDock Vina 相对 AutoDock 4 拥有更高的精度,更加推荐使用 AutoDock Vina。
注:在一些文献中,可能会将 AutoDock 4 直接称为 AutoDock,将 AutoDock Vina 称为 Vina。下文中的 AutoDock 也是指 AutoDock 4。
2 软件安装
2.1 安装软件
下载并安装 AutoDock 4 (AutoDock 主程序): http://autodock.scripps.edu/downloads
下载并安装 MGLTools 最新版本 (AutoDock 可视化程序): http://mgltools.scripps.edu/downloads
可选:
下载并安装 Open Babel(化学文件格式转换): https://github.com/openbabel/openbabel/releases/
下载并安装 PyMOL(分子可视化工具):How to install PyMOL on Windows
注:在网上的一些教程中,安装 AutoDock 需要单独安装 Python 2.5 环境,事实上 AutoDock 安装包中自带,无需单独安装。
2.2 默认路径设置
- 双击桌面“AutoDockTools-1.5.7”图标,打开“AutoDockTools”。
- 设置工作路径:依次点击“File > Preferences > Set...”,在“Set User Preferences”窗口下的“Startup Directory”更改想要设置的工作路径,并点击“Set”保存。
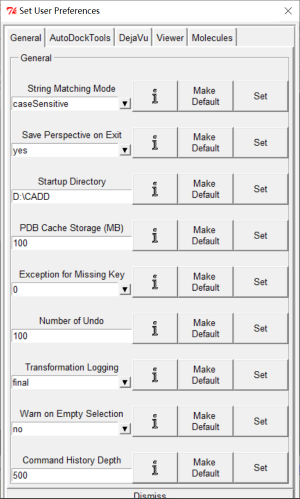
这里将工作路径设置为“D:\CADD”。
3 分子对接
首先需要对 Receptor 和 Ligand 进行预处理,并转换为 AutoDock 4 可接受的“pdbqt”文件格式。
Receptor 准备包括删除受体中的水、Ligand 等杂原子,修复残基,加氢等,这些步骤也可以使用其它软件进行操作;(Ligand 准备)接着对 Receptor 加电荷,确认可旋转键(生成不同构象),用于对接。 Ligand 若为肽也需要类似操作。(Grid map 准备)在完成 Receptor 和 Ligand 预处理后,需要进一步定义对接位点。
在拥有预处理的 Receptor 和 Ligand 及对接位点这三个信息后,就可以进行分子对接。
3.1 Receptor 准备
注:开始之前建议仔细阅读 The Protein Preparation Process,以增加对蛋白处理的理解。
打开“AutoDockTools-1.5.7”。
“File > Read Molecule”打开 Receptor 蛋白。
删除多余的原子:删除水分子和其他杂原子。
注:不是所有的水分子和杂原子都要删除,如果水分子或杂原子参与了蛋白和配体的相互作用,则应保留。
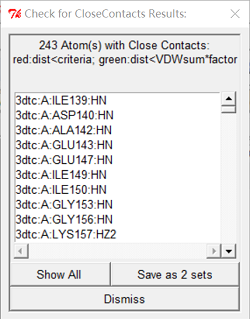
- 修复残基:“Edit > Misc > Check for Missing Atoms”,在“Check for Missing Atoms Results:”窗口中选择“Select All Residues”,点击“Dismiss”关闭窗口,再依次点击“Edit > Misc > Repair Missing Atoms”,等待出现“Check for CloseContact Results:”点击“Save as 2 sets”保存结果,点击“Dismiss”关闭窗口。
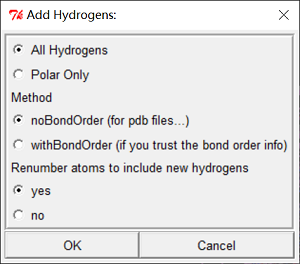
- 加氢:“Edit > Hydrogens > Add”,添加全部氢。
加电荷:“Edit > Charges > Compute Gasteiger”,点“OK”,然后点击“Edit > Charges > Check Totals on Residues”,点击“Spread Charge Deficit over all atoms in residue”,点击“Dismiss”关闭窗口。
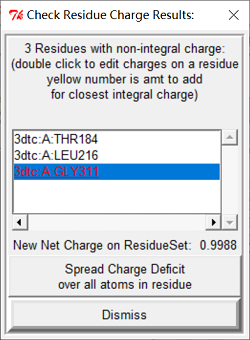
注:在 Autodock 3 官方文档中,推荐小分子添加 Gasteiger charges,蛋白质或肽添加 Kollman charges;Autodock 4官方文档中一律使用 Gasteiger charges。
合并非极性氢:“Edit > Hydrogens > Merge Non-Polar”。添加原子类型:Edit > Atoms > Assign AD4 type,点击后不出现对话框;(注:非必需,如果不执行此步骤,直接使用“File > Save > Write PDBQT”导出文件会报错)导出:“Grid > Macromolecule > Choose…”,选择正在编辑的 Receptor,提示保存 .pdbqt 文件。根据官方文档的描述,此步会自动执行上述 6 – 8 的步骤,因此只执行此步即可。
3.2 Ligand 准备
配体同样需要进行预处理,小分子使用 ChemDraw 等进行结构优化后,保存为 .mol2 格式,肽则需要按照上述“Receptor 准备”进行预处理,保存为“pdbqt”文件格式。下面以小分子配体为例:
导入 Ligand:“Ligand > Input > Open”,将文件名搜索框右侧文件格式改为MOL2 Files,打开小分子,弹出对话框点确定。
判断配体的 Root:“Ligand > Torsion Tree > Detect Root”。
设置可选键的数量:“Ligand > Torsion Tree > Set Number of Torsions”,默认选择“fewest atoms”,数值默认最大值。(此步可不操作,默认设置即可)查看可旋转的键:“Ligand > Torsion Tree > Choose Torsion”,绿色的表示可以旋转,然后点击 Done。
输出:点击“Ligand—Output > Save as PDBQT”。
3.3 Grid map 准备
- 导入 Receptor:上述 3.1.8 已经导入了 Receptor;或者直接调用保存的 Receptor 的 .pdbqt 文件,依次点击“Grid > Macromolecule > Open”,提示是否保留分子中的电荷,选择“Yes”。
导入 Ligand:“Grid > Set Map Types > Choose Ligand…”。
设置 Grid Box:“Grid > Grid Box…”,打开“Grid Options ”对话框。调整x,y,z及格子中心坐标。Spacing (angstrom)包含可以整体调整 Grid 大小。设置好 Grid Box 大小后,点击“File > Close saving current”退出。Grid 要包含大分子的活性位点,如果没有明确的活性位点,则将整个大分子包含在内。


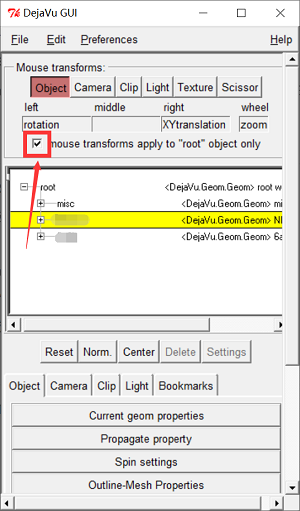
- 确认 Ligand 在 Box 外:在 3.3.2 中导入了 Ligand,如果 Ligand 在 Box 中,可能会影响对接,点击“DejaVu GUI” ,在 DejaVu GUI 窗口中选中 Ligand,取消勾选“mouse transforms apple to “root” object only”后可以单独移动 Ligand 分子,将 Ligand 移出 Box 外再勾选上“mouse transforms apple to “root” object only”。
保存:“Grid > Output > Save GPF...”,文件名需要手动加上后缀名“.gpf”。
运行:“Run > Run AutoGrid”,如下图所示,在“Program Pathname”框中选择“atuogrid4.exe”,“Parameter Filename”框中选中上一步生成的 .gpf 文件,点击 Launch。等待“Autodock Process Manager”窗口的任务完成,弹窗会自动消失,此时工作目录会多出一堆文件。

3.4 Docking 准备
导入 Receptor:“Docking > Macromolecule > Set Rigid Filename…”。
导入 Ligand:“Docking > Ligand > Choose...”,对话框选择Accept。
设置算法:“Docking > Search Parameters > Genetic Algorithm”,默认设置,点击Accept。
设置对接参数。ADT菜单栏:“Docking > Docking Parameters…”,默认设置,点击Accept。
保存:“Docking > Output > Lamarckian GA (4.2)…”,文件名需要手动加上后缀名“.dpf”。
运行:“Run > Run AutoDock”,等待弹窗的任务完成,弹窗会自动消失,对接完成。
4 结果分析
先清除显示窗口的分子:“Edit > Delete > “Delete All Molecules”。
打开 Docking 结果文件:“Analyze > Dockings > Open...”,打开“.dlg”文件。
显示 Receptor:“Analyze > Macromolecule > Open...”,显示窗口自动导入 Receptor。
查看 Docking 结果:“Analyze > Conformations > Play, ranked by energy...”。
参考文献
[2] https://autodock.scripps.edu/wp-content/uploads/sites/56/2022/04/AutoDock4.2.6_UserGuide.pdf
更新说明
2024-04-23
- 重写 1 关于 AutoDock;
- 补充部分注释。